
Kaputte Uhr brachte Wissenschaftler auf die Spur
Innsbrucker RNA-Forscher:innen klären Zusammenhänge von vier Seltenen Krankheiten
Wissenschafter im Labor von Alexander Hüttenhofer, Direktor des Instituts für Genomik und RNomik der Medizinischen Universität Innsbruck, ist es gelungen, die Rolle von nichtkodierenden RNAs bei der Entstehung des seltenen Schaaf-Yang-Syndroms zu klären. Im Zuge des Projekts entdeckten sie dabei zusätzlich die genetischen Zusammenhänge zwischen insgesamt vier Seltenen Krankheiten. Das renommierte Fachmagazin American Journal of Human Genetics veröffentlichte nun die Forschungsarbeit an der neben weiteren inneruniversitären Einrichtungen am Innsbrucker Biozentrum, die Univ.-Klinik für Pädiatrie I Innsbruck, eine Arbeitsgruppe der Leopold-Franzens-Universität Innsbruck sowie Heidelberger Forscher beteiligt waren.
Pressebilder zum Herunterladen:
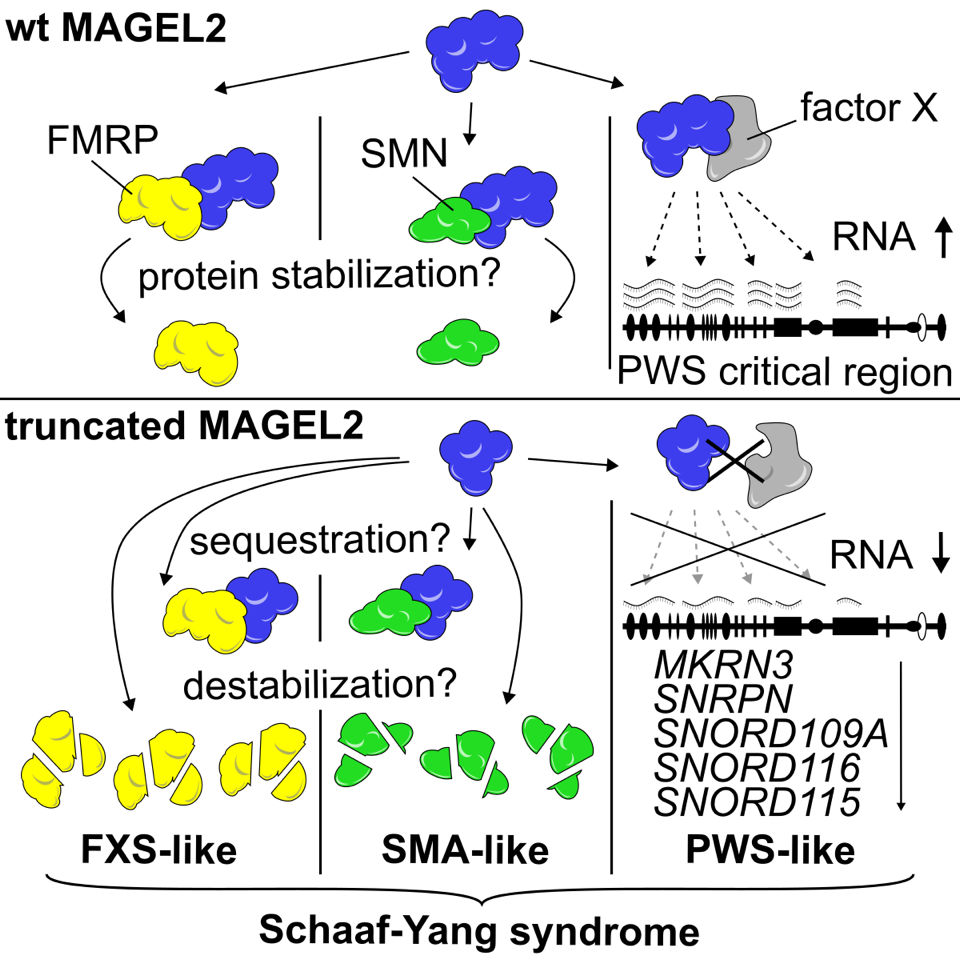
BU: Grafische Darstellung des Forschungswegs (Bild: American Journal of Human Genetics)

BU: Institutsdirektor Alexander Hüttenhofer (MUI/D. Bullock)

BU: Alexander Hüttenhofer im Labor (MUI)

BU: Erstautor David Heimdörfer und Institutsdirektor Alexander Hüttenhofer (MUI)
Innsbruck, 25. Juni 2024: Vor sieben Jahren ging Alexander Hüttenhofer, dem Direktor des Instituts für Genomik und RNomik an der Medizinischen Universität Innsbruck, seine Armbanduhr richtig auf den Zeiger. Heute kann er nun behaupten, dass ihn der unzuverlässige Zeitmesser, der oft kaputt war, zur genetischen Klärung von vier Krankheitsbildern geführt hat. Bei den vielen Besuchen in dem Tiroler Geschäft wegen Reparatur seiner Uhr kam er irgendwann mit dem Uhrmacher ins Gespräch, der ihm von seinem Kind erzählte. Die damals etwa acht Jahre alte Tochter konnte nicht laufen, sie sprach nicht und zeigte Symptome von Autismus. Diese Eigenschaften ließen Hüttenhofer hellhörig werden. Sie erinnerten ihn zum Teil an Patient:innen mit dem Prader-Willi-Syndrom.
Wie es der Zufall wollte, war Hüttenhofer im Jahr 2000 in Münster als Projektleiter am Deutschen Humanen Genom Projekt beteiligt, bei dem sich seine Arbeitsgruppe erstmals mit der Identifizierung von nicht-Protein-kodierenden RNAs (ncRNA) auseinandersetzte. Heute weiß man, dass ncRNAs mit ca. 96 Prozent im Vergleich zu den proteinkodierenden Genen (d.h. messenger RNAs) mit ca. vier Prozent die überwältigende Mehrheit an RNA in menschlichen Zellen ausmachen. Eine der Hauptaufgaben der ncRNAs ist dabei die Genregulierung: ncRNAs schalten einzelne Gene ein oder aus und regeln damit deren Proteinproduktion. Im Zuge des Humanen Genom Projekts entdeckte Hüttenhofer damals drei ncRNAs, die überraschenderweise nur im Gehirn angeschaltet wurden und die er HBII-13, HBII-52 und HBII-85 nannte. „Bis dahin dachte man, dass ncRNAs in allen Zellen dasselbe machen. Diese drei nichtkodierenden RNAs, die auf einem Abschnitt des Chromosom 15 des Menschen kodiert sind, waren damit die ersten identifizierten ncRNAs im Gehirn“, schildert er.
Vom Prader-Willi-Syndrom zum Schaaf-Yang-Syndrom
Wiederum einige Jahre später, 2008, identifizierten Wissenschafter:innen die genetische Ursache des Prader-Willi-Syndroms (PWS), das bei 1:15.000 Neugeborenen auftritt. Den Betroffenen mit PWS fehlt meistens ein ganzer Chromosomenabschnitt (d.h. vier Millionen Basenpaare) auf dem väterlichen Chromosom 15 und interessanterweise damit auch die Gene für die ncRNAs HBII-13, HBII-52 und HB-85. Noch spannender war, dass sich später bei weiteren PWS Patient:innen herausstellte, dass ihnen nur die inzwischen in SNORD116 umbenannte HBII-85 RNA, die als sogenanntes Gencluster in insgesamt 30 Genkopien auf dem Chromosom 15 vorkommt, fehlte und dieses Fehlen unmittelbar für die PWS-Symptomatik verantwortlich ist. „Das war das erste Mal, dass man gesehen hat, dass eine ncRNA auch eine Krankheit verursachen kann“, erklärt Alexander Hüttenhofer. Kinder mit PWS haben Bewegungsschwierigkeiten (muskuläre Hypotonie), sind in der mentalen Entwicklung verzögert und sie spüren kein Sättigungsgefühl, was zu schwerem Übergewicht und entsprechenden Folgeerkrankungen führt.
Als Hüttenhofer Jahre später vom Uhrmacher die Beschreibung der Symptome der Tochter hörte, überzeugte er mit Unterstützung des Genetikers Andreas Janecke und Kinderklinik-Direktor Thomas Müller die Eltern, das Kind genetisch untersuchen zu lassen. „80 Prozent der Eigenschaften der Tochter haben mich an das Prader-Willi-Syndrom erinnert, aber sie stimmten nicht ganz damit überein. In Peking haben wir dann die Genome der Tochter und beider Eltern sequenzieren lassen.“ Bei der Tochter, nicht aber bei den Eltern, wurde eine Mutation im sogenannten MAGEL2 Proteingen gefunden, welches zu ähnlichen Symptomen wie PWS führte und das dabei ebenfalls auf Chromosom 15 in unmittelbarer Nähe zum SNORD116 Cluster lag.
Bei der Tochter fehlte also nicht das SNORD116 Gencluster selbst, wie beim Prader-Willi-Syndrom, sondern es gab eine Mutation im MAGEL2 Gen. Die MAGEL2 Mutation war zuvor bereits als Verursacher des „Prader-Willi-ähnlichen Syndroms“ bei weltweit etwa nur 120 Kindern mit ähnlichen Symptomen wie PWS beschrieben und wurde später nach seinen Entdeckern als Schaaf-Yang-Syndrom (SYS) bezeichnet. „Andreas Janecke hat herausgefunden, dass bei der Patientin wirklich nur ein einziges Nukleotid zusätzlich vorhanden ist, welches die Funktion des MAGEL2 Proteins stört“, erklärt Hüttenhofer. Das Ziel seiner Arbeitsgruppe war es nun, den Zusammenhang zwischen dem mutierten MAGEL2-Gen und dem Fehlen der SNORD116 RNA herzustellen, die beide eine sehr ähnliche Symptomatik verursachten. Sie konnte zeigen, dass nur ein funktionales MAGEL2 Protein in der Lage war, die SNORD116 RNA in ausreichenden Mengen zu produzieren. „Wenn es aber mutiert ist, kann es nur eine sehr geringe Menge an SNORD116 erzeugen. Dies kann, zumindest teilweise, die ähnliche Symptomatik zwischen den Krankheiten PWS und SYS erklären“, so der Wissenschaftler. Die Familie des Mädchens mit SYS kennt jetzt den zu erwartenden Verlauf der Erkrankung. Aufgrund von anderen Patient:innen mit SYS wissen die Eltern nun, dass die Tochter nichts von dem, was sie einmal dank Fördermaßnahmen wie Physiotherapie gelernt hat, wieder verlernen wird. Eine ursächliche Behandlung für SYS Patient:innen gibt es zwar bisher nicht, allerdings könnten die vorliegenden Forschungsergebnisse zu neuen Therapieansätzen führen.
Zusammenhang mit Spinaler Muskelatrophie und Fragile-X-Syndrom
Für das Team um Hüttenhofer, der in dem Projekt neben der Kollaboration mit der Univ.-Klinik für Pädiatrie I eng mit Kolleg:innen vom Innsbrucker Biozentrum, d.h. dem Institut für Zellbiologie (Direktor: Lukas Huber), dem Institut für Pathophysiologie (Direktor: Hesso Farhan), der Core Facility Proteomics am Institut für Medizinische Biochemie (Direktor: Ludger Hengst), und dem Stammzellenforscher Frank Edenhofer vom Institut für Molekularbiologie der Universität Innsbruck, sowie dem Heidelberger Humangenetiker Christian Schaaf – einer der Namensgeber des Schaaf-Yang-Syndroms – zusammenarbeitete, waren damit noch nicht alle Rätsel gelöst. Sie machten sich auf die Suche nach weiteren Interaktionspartnern des MAGEL2 Proteins, das die gesamte Symptomatik des Schaaf-Yang-Syndroms erklären konnte. Im Laufe des Projekts entdeckten sie dabei Zusammenhänge der MAGEL2 Mutation zu zwei weiteren Seltenen Krankheiten, die dem SYS ähneln: dem Fragile-X-Syndrom (FXS), das unter anderem als Symptomatik auch Autismus zeigt und der Spinalen Muskelatrophie (SMA), bei der die Patient:innen sukzessive ihre Bewegungsfähigkeit verlieren. Beide Symptomatiken werden auch bei Schaaf-Yang-Syndrom Patient:innen beobachtet. Damit konnte also, ausgehend von einer einzigen ncRNA und einer Mutation im MAGEL2 Gen, der Zusammenhang zwischen vier unterschiedlichen Seltenen Erkrankungen hergestellt werden.
Erstautor der nun im American Journal of Human Genetics publizierten Studie ist David Heimdörfer, der im Rahmen des Projekts seine Doktorarbeit abschließen konnte. Alexander Vorleuter, der als Co-Erstautor firmiert, absolvierte damit sein Masterstudium. Institutsdirektor Alexander Hüttenhofer freut sich insbesondere über die erfolgreichen Kooperationen durch die es gelungen ist, die Bedeutung der RNA-Forschung an der Medizinischen Universität Innsbruck weiter hervorzustreichen und vier Seltene Krankheiten besser zu verstehen. Seit Beendung der Studie tickt Hüttenhofers Uhr übrigens ohne jegliche Probleme und ziert mittlerweile das Armgelenk seines Sohnes.
Forschungsarbeit:
David Heimdörfer et al, Truncated variants of MAGEL2 are involved in the etiologies of the Schaaf-Yang and Prader-Willi syndromes,The American Journal of Human Genetics, 2024, ISSN 0002-9297, https://doi.org/10.1016/j.ajhg.2024.05.023.
PR & Medien
Medienkontakt:
Medizinische Universität Innsbruck
Public Relations und Medien
Theresa Mair
Innrain 52, 6020 Innsbruck, Austria
Telefon: +43 512 9003 71833
public-relations@i-med.ac.at, www.i-med.ac.at