
16.02.2024
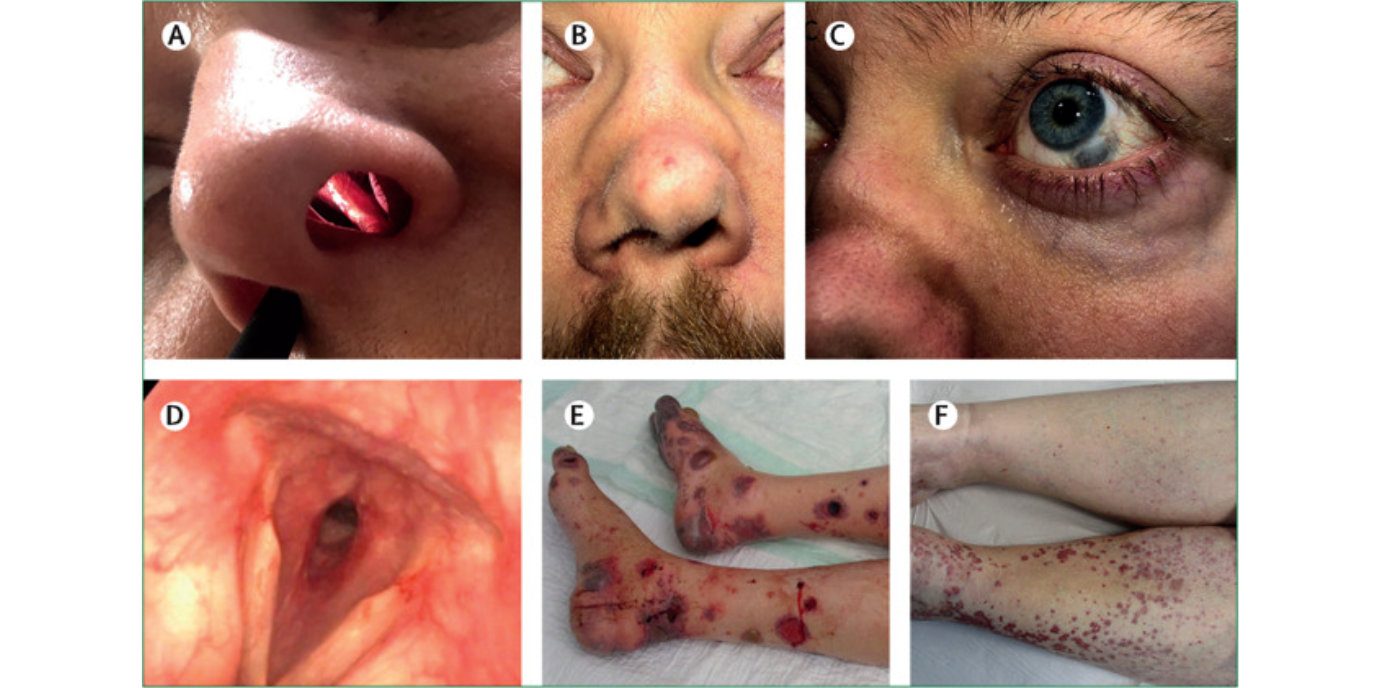
Große Fortschritte für Vaskulitis-Patient:innen
130 Referenzen, davon ganze 60 Publikationen seit dem Jahr 2020, sind in die Übersichtsarbeit über die ANCA-assoziierte Vaskulitis eingeflossen, die Andreas Kronbichler (Univ.-Klinik f. Innere Medizin IV) soeben mit vier internationalen Kolleg:innen im hochrangigen Fachjournal The Lancet veröffentlichen konnte. Das ist außergewöhnlich. Denn bei ANCA-assoziierten Vaskulitiden handelt es sich um Seltene Krankheiten.
Bei der ANCA-assoziierten Vaskulitis handelt es sich um eine systemische Autoimmunerkrankung, die eine Entzündung der kleinen Gefäße hervorruft. „Von den ersten Symptomen bis zur Diagnose dauert es im Durchschnitt drei Monate“, sagt Andreas Kronbichler von der Univ.-Klinik für Innere Medizin IV (Nephrologie, Direktor: Gert Mayer). Ein Grund für die verzögerte Diagnose ist, dass die Krankheitszeichen anfangs diffus sind: Nachtschweiß, Gewichtsverlust, Muskelschmerzen gehören dazu. Erst im Verlauf werden die Symptome organspezifisch – von Nasenbluten, neuropathischen Schmerzen, Blut im Harn bis Bluthusten. „Die Entzündung frisst das Gewebe weg und war früher sehr entstellend. Vom zentralen Nervensystem bis zum peripheren Nervensystem können alle Organe betroffen sein. Die Erkrankung kann grundsätzlich in jedem Alter auftreten, wobei der Gipfel zwischen 50 und 70 Jahren liegt. Wenn Kinder an ANCA-Vaskulitis erkranken, sind allerdings zwischen 70 und 80 Prozent weiblich“, erklärt der Spezialist. Im Ewachsenenalter sind Frauen und Männer zum gleichen Ausmaß betroffen.
Ein weiterer Grund für die späte Diagnose ist die Tatsache, dass die ANCA-assoziierte Vaskulitis eine Seltene Krankheit ist, welche wegen der unterschiedlichen Organmanifestationen die Aufmerksamkeit und die Erfahrung von Mediziner:innen vieler Disziplinen bedarf. Innsbruck ist auf diesem Gebiet „weit vorne, unter den Top 10 weltweit“, sagt Kronbichler. Er schätzt, dass jährlich in Tirol pro Bezirk 2,5 bis drei Patient:innen neudiagnostiziert werden. Die Inzidenz liegt bei 25 zu einer Million. Im Moment sind 80 Betroffene in Innsbruck in Behandlung. Kronbichler geht von einer hohen Dunkelziffer aus. „Die Diagnose an sich ist nicht schwierig, wenn die Erfahrung da ist. Die Patient:innen brauchen jemanden, wo sie hingehen können“, erklärt der Spezialist, der erst vor kurzem von einem 18-monatigen Aufenthalt in Cambridge zurückgekehrt ist.
Gemeinsam mit Kolleg:innen aus Schweden, Brasilien, den USA und den Niederlanden hat er den Stand der Forschung bezüglich Therapie und Diagnose zusammengetragen. Es hat sich viel getan in den vergangenen drei Jahren – obwohl Seltene Krankheiten entsprechend selten im Fokus des weltweiten Forschungsinteresses liegen. „Die Europäische Vaskulitis Gesellschaft (EUVAS) gibt es seit 30 Jahren. Die Kommunikation ist fantastisch und sehr kollaborativ. Wir haben auch die Pharma für das Thema begeistern können“, sagt Kronbichler und erklärt damit das große Interesse und Vorankommen der Expert:innen-Community in der Vaskulitis-Forschung. „Es sind schon neue Studien in Planung.“
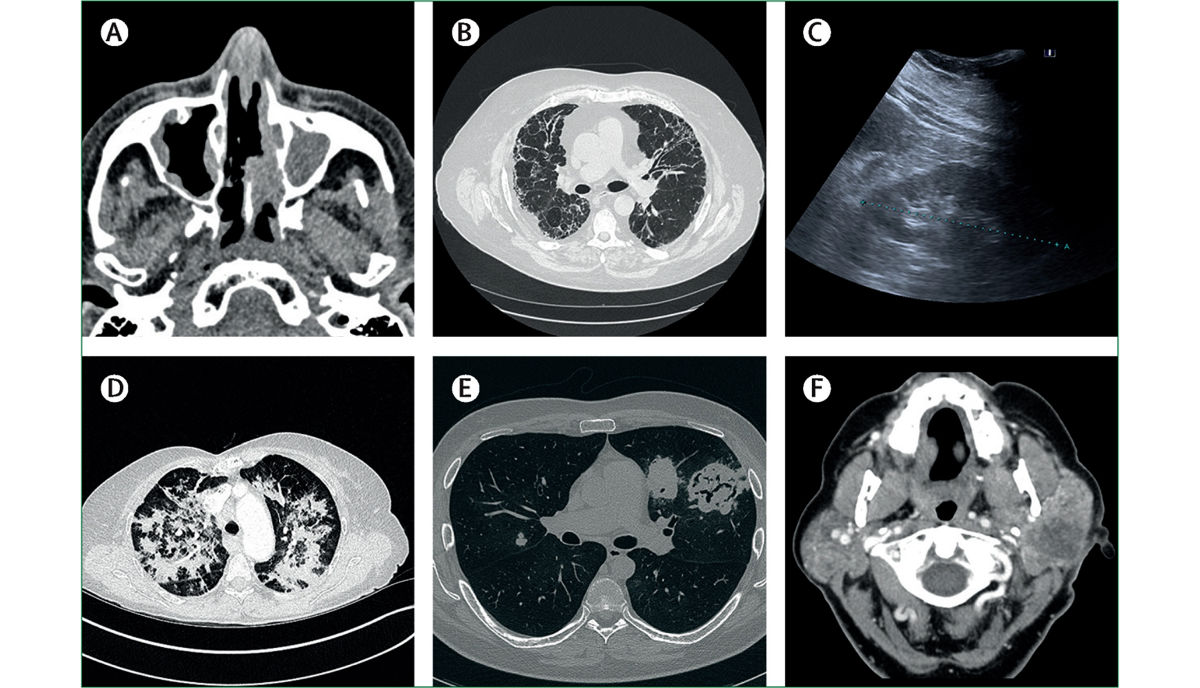
BU: In der Bildgebung zeigen sich Manifestationen von ANCA-assoziierter Vaskulitis
Das diagnostische “State of the art”-Prozedere sieht, wie im Paper beschrieben, zunächst die Erhebung von Blutlabor, allen voran des CRP-Werts und der Blutsenkungsgeschwindigkeit, vor. „Eine Senkung von ≥ 50 Milliliter pro Stunde spricht oftmals für eine systemische Autoimmunerkrankung“, erklärt Kronbichler. In der Folge werden andere Untersuchungen (z.B. CT und MRT, je nach Organbefall) angeordnet. „Das Wesentliche um eine Vaskulitis endgültig zu diagnostizieren, ist aber die Biopsie des symptomatischen Organs“, sagt er. Häufig würden dabei Granulome, also kugelförmige Ansammlungen von Entzündungszellen, oder vaskulitische Manifestationen (v.a der Niere) entdeckt. Anhand der Gewebeprobe können zudem Faktoren für die Prognose bestimmt werden.
Sehr viel getan hat sich zuletzt bei den therapeutischen Möglichkeiten, insbesondere bei den zwei Erkrankungsformen Granulomatose mit Polyangiitis (GPA) und mikroskopischer Polyangiitis (MPA), die in der Publikation berücksichtigt wurden. Für beide gibt es jeweils einen spezifischen Bluttest – den Protenase-3-ANCA und den Myeloperoxitase-ANCA-Test. 2020 und 2021 sind dazu Arbeiten im New England Journal of Medicine erschienen.
Dank innovativer Therapien, wie z.B. dem Komplementhemmer Avacopan (C5a Rezeptor 1-Inhibitor) ist es inzwischen möglich, bei GPA und MPA die bisherige kumulative Steroidtherapie, frühzeitig auszuschleichen und die damit verbundenen Nebenwirkungen (u.a. Infektanfälligkeit, Diabetes) hintanzuhalten. Zusätzlich oder alternativ zu dem Komplementhemmer sollte außerdem Rituximab, ein Antikörper gegen CD20-tragende Zellen, verabreicht werden. „Nach einer sechsmonatigen Einleitungstherapie folgt analog zu den Empfehlungen eine 18- bis 42-monatige Erhaltungstherapie. Bei hohem Rückfallrisiko wird die Therapie fortgesetzt“, erläutert Kronbichler. Die Patient:innen, die zu Therapiebeginn meist in schlechtem Zustand seien, würden „dank des besseren Verständnisses der Erkrankung und der neuen Therapien“ mittlerweile im Verlauf der Behandlung großteils von einer nahezu normalen Lebensqualität berichten. Die Forschung, zur Verbesserung des Lebens von Patient:innen mit ANCA-assoziierter Vaskulitis wird aber noch lange nicht ruhen: „Es gibt Bestrebungen, mit einer Car-T-Zell-Therapie das Immunsystem quasi umzuprogrammieren, um damit eine langanhaltende Remission erzielen zu können“, gibt Kronbichler einen Ausblick auf die Zukunft.
(Innsbruck, 16.2.2024, Text: T. Mair, Bilder: ElSevier/The Lancet (2), privat)
Zur Forschungsarbeit: Andreas Kronbichler, Ingeborg M Bajema, Annette Bruchfeld, Gianna Mastroianni Kirsztajn, John H Stone, Diagnosis and management of ANCA-associated vasculitis, The Lancet, Volume 403, Issue 10427, 2024,Pages 683-698, ISSN 0140-6736,
https://doi.org/10.1016/S0140-6736(23)01736-1.

Zur Person: Andreas Kronbichler hat in Innsbruck Humanmedizin studiert und ist an der Univ.-Klinik für Innere Medizin IV (Nephrologie) tätig, wo er sich schwerpunktmäßig mit Autoimmunität und Nierenerkrankungen beschäftigt. Von 2014 bis 2015 sowie von 2021 bis 2023 war er am Addenbrooke’s Hospital bzw. an der University Cambridge tätig. Als erst zweiter österreichischer Arzt ist er 2023 mit dem Nils-Alwall-Preis der Deutschen Gesellschaft für Nephrologie ausgezeichnet worden. 2021 erhielt er den Otto-Kraupp-Preis für die beste Habilitation.
Weitere Links:
Universitätsklinik für Innere Medizin IV (Nephrologie), Innsbruck
mypoint-Bericht: Innsbrucker Forschungsarbeit verbessert Therapie von seltener Gefäßerkrankung
mypoint-Bericht: Systemische Vaskulitiden – eine interdisziplinäre Herausforderung
mypoint-Bericht: Otto-Kraupp-Preis 2021 für Andreas Kronbichler
mypoint-Bericht: Dr. Otto Seibert-Preis 2019 für Andreas Kronbichler