
Research Group Hereditary cancer genetics/Group Wimmer
Principal Investigator: Ao. Univ.-Prof. Dr. Katharina Wimmer (CV, Publications)
Institut of Human Genetics
Medical University of Innsbruck
Peter-Mayer-Str. 1
6020 Innsbruck / Austria
+43 (0)512-9003-70513
katharina.wimmer@i-med.ac.at
Mission Statement
The research group Hereditary Cancer Genetics works in close collaboration with the diagnostics laboratory and aims at improving the diagnostics and management of patients and their families with tumour predisposing disorders. The group focusses on two hereditary tumour syndromes, neurofibromatosis type 1 (NF1) and constitutional mismatch repair-deficiency (CMMRD), that show clinical overlap.
Main goals are:
- development of new approaches improving the sensitivity and specificity of molecular diagnostics of NF1, CMMRD and other hereditary tumour syndromes
- understanding how genetic alterations found in patients with hereditary tumour syndromes interfere with gene processing, specifically exon definition in mRNA splicing
- delineating the clinical presentation of CMMRD and understanding the correlation between genotype and phenotype
- assessing the prevalence of CMMRD in paediatric cancer patients
- uncovering the genetic mechanisms underlying the development of neoplastic and non-neoplastic features in CMMRD patients
- delineating the genetic cause of CMMRD-like phenotypes
Background Information about our scientific projects:
Neurofibromatosis type 1 (NF1)
NF1 is a common condition affecting ~1/3000 individuals worldwide, with half of the patients being sporadic. NF1 is a progressive disorder with more signs developing with time. Hallmark features of NF1 are multiple (>6) café-au-lait macules (CALMs), skinfold freckling, dermal and plexiform neurofibromas, Lisch nodules, optic pathway glioma and specific bone abnormalities. The spectrum of NF1 mutations is very broad. NF1 is notorious for its phenotypic variability. Even within a family, the phenotype can vary markedly between affected individuals with the same NF1 mutation (genotype). In general, there is little correlation between the genotype and the phenotype in NF1. Nevertheless, due to the broad application of genetic NF1 diagnostics with highly sensitive mutation analysis protocols, as they are used in our diagnostics laboratory, and a systematic recording of the associated phenotypes, a constantly increasing number of mutations are identified that are recurrently associated with specific clinical NF1 phenotypes. It is a long-term goal of our laboratory to contribute to the delineation of further genotype-phenotype correlations that have major implications for the management and counseling of the patients.
The variant spectrum of NF1 mutations is very complex and includes a wealth of sequence variants affecting mRNA splicing. The RNA-based mutation analysis protocols used in our diagnostics laboratory effectively detect also non-canonical splice mutations. This increases not only mutation detection rates, but also provides us with a unique data set that can be used to study the mechanisms of action of non-canonical splice mutations. This in turn helps to gain insights into cellular processes by which splice sites are recognized and selected and has immediate practical implications for molecular genetic diagnostics of NF1 and beyond, as it leads to improved algorithms to stratify which gene variants of unknown significance (VUS) are likely to have a splice effect and, hence, are of clinical importance. See e.g. Wimmer et al. 2020, Human Mutat 41:1145–1156.
Constitutional Mismatch Repair Deficiency (CMMRD)
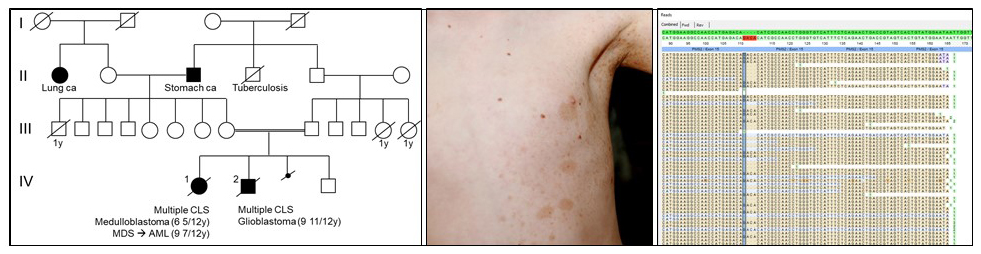
CMMRD is caused by bi-allelic mutations in one of the four DNA mismatch repair (MMR) genes, MLH1, MSH2, MSH6 and PMS2. This rare condition (estimated incidence: one in a million children from unrelated parents) was recognized as a distinct childhood cancer susceptibility syndrome in 1999 (Ricciardone et al. 1999 Cancer Res, 59:290–3 and Wang et al. 1999, Cancer Res 59:294–7). CMMRD is associated with an extraordinarily high tumor risk. The spectrum of CMMRD-associated malignancies is very broad, including primarily hematological malignancies, tumors of the central nervous system, and carcinomas associated with Lynch syndrome.
Over 50% of CMMRD patients have PMS2 mutations, a gene that is a recognized “dead zone” for genomic sequencing due to its multiple pseudogenes. Therefore, we established mutation analysis protocols that allow reliable and effective mutation analysis also in this MMR gene. By stating a molecular diagnosis in a number of patients, our laboratory substantially contributed to the delineation of the tumor spectrum and non-neoplastic features that serve as signposts of the condition in a pediatric cancer patient. This led to the development of criteria for the clinical suspect diagnosis in pediatric cancer patients proposed by a European consortium Care for CMMRD (C4CMMRD).
CMMRD shows clinical overlap with neurofibromatosis type 1 (NF1) (for review see Wimmer, Rosenbaum, Messiaen 2017, Clin Genet 91:507-519). Hence, CMMRD is a possible differential diagnosis in suspected sporadic NF1 children for whom no NF1 or SPRED1 mutation is identified. Using a newly developed screening assay for CMMRD (Gallon et al. 2019, Hum Mutat 40:649-655 we show that the prevalence of CMMRD is 0.41% among these children (Perez-Valencia et al. 2020, Genet Med, Online ahead of print). This confirms estimations on which C4CMMRD guidelines are based that advocate CMMRD testing of preselected children rather than reflex testing of all suspected sporadic NF1 children lacking NF1/SPRED1 mutations.