
Human Genetics
Research Focus
General Facts
Research
Selected Publications
Selected Funding, Collaboration
Keywords: Human Genetics, Biochemical Genetics, Inherited metabolic disorders, Lipidomics, Inherited tumour disposition syndromes, Tumour genetics, Ehlers-Danlos syndromes, Dental genetics, Neurogenetics, Reproductive Medicine, Syndromology
Research (ÖSTAT Classification): 301301, 301303, 301304, 304002
Research Focus
- Genetic causes of rare diseases and syndromes, incl. inherited metabolic diseases, Ehlers-Danlos syndromes, dental genetics, neurogenetics, reproductive genetics, developmental disorders and others.
- Genetic causes of tumours and tumour dispositions, incl. hereditary cancer disposition syndromes, hamartomatous tumours, haematological malignancies and melanoma.
- Membrane lipid metabolism of organelles and cells.
- Transcription and transcript processing, splicing.
- New genetic laboratory methods, genotyping arrays, methylome analysis, mutation databases, quality control.
- Genetic counselling.
General Facts
The primary aim of the Institute of Human Genetics is to clarify the genetic determinants of health and disease in humans and to translate this knowledge into individual medical care. This aim is achieved by combining comprehensive clinical services and all relevant laboratory diagnostic techniques with basic research. The main focus is on rare diseases that are inherited as monogenic traits, with expansion to polygenic disease risk factors. The institute includes the Innsbruck Centre for Medical Genetics, which provides medical genetic services and genetic counselling for Western Austria, with extensive outpatient clinics and in-patient consultation in Innsbruck as well as several regional hospitals. The diagnostic and research laboratories cover all the relevant methods of molecular genetic (DNA, RNA, epigenetics) and cytogenetic (genomic) analysis, biochemical analysis with a focus on lipidomics, and a wide range of functional and cell biology techniques. The institute is dedicated to interdisciplinary collaboration and shares research activities with a large number of cooperation partners.
Research
Biochemical Genetics
Johannes Zschocke, Markus Keller
The characterisation of the clinical, biochemical and molecular features of inherited metabolic disorders is a longstanding research focus of the institute. Areas of special interest include lipid, lipoprotein, fatty acid, organic acid, amino acid, dipeptide and polyamine metabolism, as well as ongoing smaller projects and publications. In addition to a range of research projects, we have led the development of the International Classification of Inherited Metabolic Disorders (ICIMD, see www.icimd.org), a group-based collation of >1400 monogenic diseases, endorsed by all international societies for inherited metabolic diseases (see Fig. 1). Another major effort in 2020 was the new, 5th edition of the Vademecum Metabolicum, the most widely used handbook on the diagnosis and treatment of inherited metabolic disorders worldwide. Previous editions of this book have been translated into >10 languages.
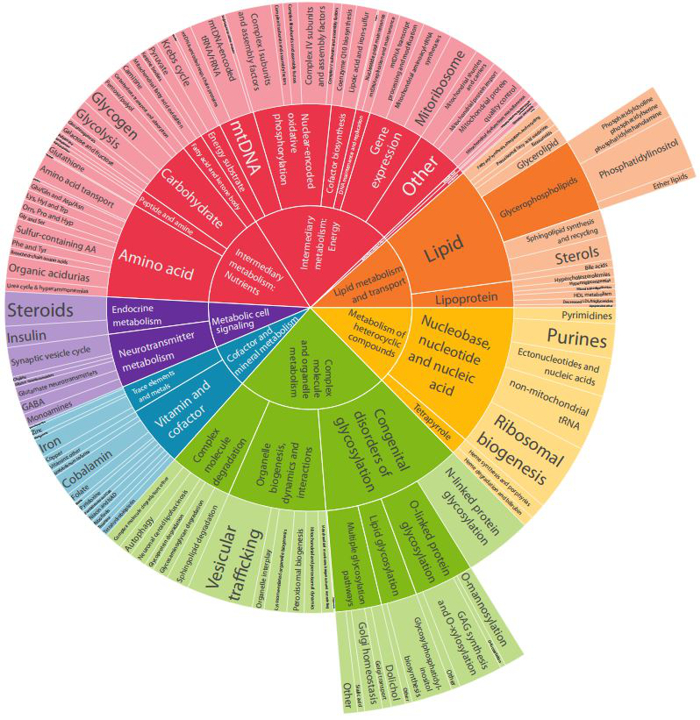
Fig. 1: Sunburst diagram of the hierarchical group structure of 1450 inherited metabolic disorders. Diagram from Ferreira C. et al., An international classification of inherited metabolic disorders, ICIMD). J Inherit Metab Dis. 2021, in press.
Biochemical Genetics Laboratory
Markus Keller
The core mission of the Biochemical Genetics Laboratory is functional elucidation of the pathobiochemical mechanisms that underlie genetically determined and predisposed diseases, by merging comprehensive genetic and multiomics data using specialist bioinformatics concepts, with the aim of also developing novel therapeutic interventions.
With the enormous possibilities of modern genetics, comprehensive functional elucidation of the pathomechanisms of genetic variants in interaction with the environment will be one of the greatest challenges for research in the coming decades. Countless cellular metabolic processes influence the development, characteristics and treatability of a wide variety of pathologies. Numerous monogenic diseases have been identified through biochemical abnormalities, and regular monitoring of specific metabolites has a central function in the management of various rare metabolic diseases. In recent years, this task has shifted to biochemical confirmation of genetically caused diseases, through assessment of specific metabolic changes linked to genetic variants identified by genome-wide analysis.
Metabolic medicine has progressed from identification of individual substances and examination of specific groups of metabolites to comprehensive analysis methods that record a large number of different substances with high sensitivity and specificity in an undirected manner (metabolomics, lipidomics). These are essential additions to genome-wide genetic analysis methods of studying the causal links between genotypes and phenotypes and they therefore foster a deeper functional understanding of molecular medicine as well as facilitating the direct integration of biochemical findings into clinical medicine, with regard both to monogenic metabolic diseases and to multifactorial conditions. In recent years, we have developed strong expertise particularly in the field of lipidomics, examining the effects of metabolic diseases on the composition of membrane lipids (e.g. in the mitochondria) and the underlying enzymatic processes.
Innovative lipidomics approaches have allowed us to study mitochondrial and cellular phospholipid metabolism in unparalleled detail and comprehensively to characterise the pathomechanisms of relevant diseases, such as Barth Syndrome. The combination of biochemical analytical tools with transcriptomics data and the use of integrative data analysis strategies as well as artificial neural network-based machine learning has clarified the remodelling process of mitochondrial phospholipids, which is impaired in Barth Syndrome patients (see Fig. 2). This focus has recently been strengthened through a shared investment of the Institute of Human Genetics and the Medical University of Innsbruck in a timsTOF Pro mass spectrometer (Bruker), which is dedicated to basic research into membrane lipid metabolism, patient-oriented biomarker and molecular signature studies, and to collaborative projects within this research area.
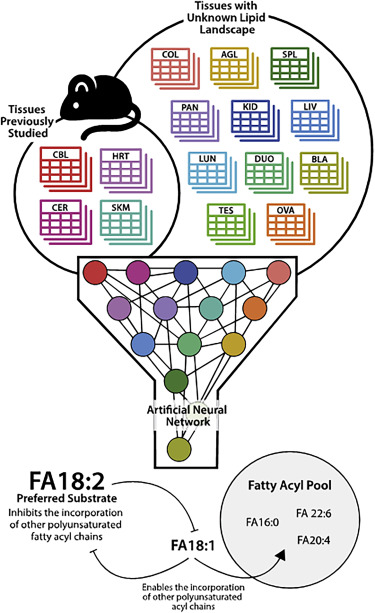
Fig. 2: Tissue-specific phospholipid acyl chain availability can be used to predict the cardiolipin landscape using artificial neural networks. Taken from the feature article by Sniezek OL et al., Cell Rep. 2020; 30: 3949 – 3950 in our publication Oemer G et al., Phospholipid Acyl Chain Diversity Controls the Tissue-Specific Assembly of Mitochondrial Cardiolipins. Cell Rep. 2020; 30: 4281.
Familial Hypercholesterolaemia
Martina Witsch-Baumgartner, Johannes Zschocke
We are interested in the genetic basis of Familial Hypercholesterolaemia (FH) in Tyrol and Austria and in the functional impact of genetic factors on disease severity and phenotype. In addition to the known monogenic causes, we are investigating polygenic causes of hypercholesterolaemia in a cohort of >300 well characterised individuals, in cooperation with the University Hospital for Internal Medicine I at MUI. The identification of polygenic risk scores (PRS) has been established using the DNA array technology (Illumina iScan) in the institute in close collaboration with the Institute of Genetic Epidemiology at MUI. We hope that these studies will further optimise therapeutic approaches, lifestyle counselling and risk assessment with regard to cardiovascular events.
Tumour Genetics
Hereditary Cancer Genetics
Katharina Wimmer
This research laboratory is strongly associated with the institute’s diagnostics laboratory, which offers molecular investigations for a broad spectrum of cancer susceptibility syndromes. We have longstanding experience with RNA-based assays for the identification of splice mutations. Current research aims to elucidate the mechanisms by means of which sequence variants alter mRNA splicing, with the ultimate goal of better understanding of the mechanisms of exon definition. Evaluation of non-canonical intronic NF1 gene 3′ splice site mutations has revealed three major motives of action of these mutations. Testing for these motives of action allows a high-precision distinction between probably benign and probably spliceogenic variants of unknown significance, which are found by gDNA testing. In addition to clinical implications, our findings lay the foundations for further studies to reveal the processes involved in 3′ splice site recognition and selection by splice mechanism.
A further research focus is on the genetic and clinical characterisation of constitutional mismatch repair deficiency (CMMRD), a rare childhood cancer predisposition syndrome caused by bi-allelic mutations in one of the four DNA mismatch repair (MMR) genes, MLH1, MSH2, MSH6 and PMS2. We contributed substantially to delineation of the tumour spectrum and non-neoplastic features that serve as signposts of the condition in a paediatric cancer patient. This resulted in the development of criteria for the clinical suspect diagnosis in paediatric cancer patients, as proposed by European consortium Care for CMMRD (C4CMMRD) under our lead. CMMRD shows a clinical overlap with neurofibromatosis type 1 (NF1), a much more frequent condition, and we investigated the frequency of CMMRD in the NF1 differential diagnosis. Using a novel CMMRD assay developed at Newcastle University in collaboration with us, we tested over 700 DNA samples from NF1 mutation-negative children and we identified CMMRD prevalence of 0.41% in this cohort. This supports current C4CMMRD consortium guidelines, which advocate CMMRD testing only of preselected children, rather than reflex testing of all suspected sporadic NF1 children who lack NF1 mutations. Our next aim is to evaluate the C4CMMRD criteria for preselection of potentially affected children in a multi-centre prospective study.
Haematological Malignancies, Melanomas
Emina Jukic
We have established a new research group with special focus on genetic and epigenetic alterations in haematological malignancies and specific solid tumours. In collaboration with the Austrian Myeloma Registry and Innsbruck University Hospital for Internal Medicine V, we are investigating the influence of genetic variants on the observed heterogeneity of multiple myeloma (MM), a heterogeneous B-cell malignancy that can be divided into clinically relevant subgroups on the basis of genetic features (see Fig. 3).
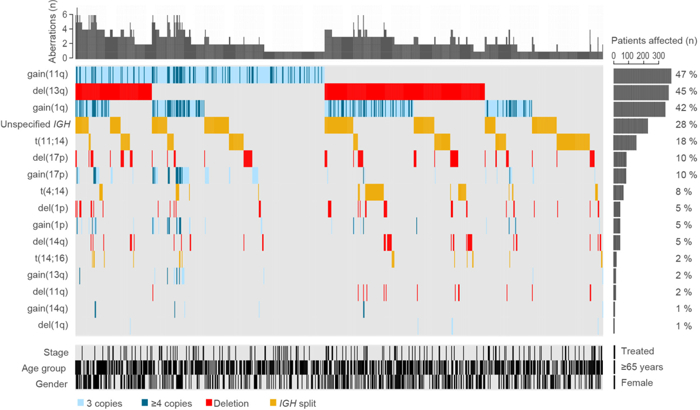
Fig. 3: Landscape of chromosomal abnormalities in 794 myeloma patients. Diagram from Locher Maurus et al., The prognostic value of additional copies of 1q21 in multiple myeloma depends on the primary genetic event. Am J Hematol. 2020; 95: 1562 – 1571.
DNA methylation is an important epigenetic mechanism for the regulation of gene activity. Cancer cells usually have altered DNA methylation patterns, which can be determined in a genome-wide fashion by means of methylome analysis, within the institute. In collaboration with the Institute of Medical Genetics and Pathology at Basel University, the Institute of Human Genetics at Bonn University and Innsbruck University Hospital for Internal Medicine V, we are studying epigenetic alterations in myeloid neoplasms, e.g. myelodysplastic syndrome. Many recurrently mutated genes in these diseases are regulators of DNA methylation. We are investigating whether methylome analysis can provide clinically useful information for the diagnosis, stratification and treatment of affected patients. In collaboration with the University Hospital for Dermatology, Venereology and Allergology at MUI, we are also assessing genetic and epigenetic – including methylome – analyses in malignant melanoma for more precise disease characterisation and to distinguish malignant lesions from benign melanocytic nevi.
Clonal haematopoiesis of indeterminate potential (CHIP) reflects the increased expansion of a cell clone in the blood, without leading to malignant disease. These cells have acquired growth-enhancing somatic mutations as a consequence of aging, exposure to environmental mutagens and other factors such as oxidative stress and DNA damage. Considering the link with inflammatory processes, we are currently studying the correlation between CHIP-associated mutations and inflammation in patients with renal diseases, in collaboration with Innsbruck University Hospital for Internal Medicine IV.
Periodontal Ehlers-Danlos Syndrome and Other Inherited Connective Tissue Disorders
Johannes Zschocke, Albert Amberger
Ehlers-Danlos syndromes (EDS) are a group of inherited connective tissue disorders characterised by joint hypermobility, skin hyperelasticity and the fragility of various tissues. We have a long track record in the identification and functional characterisation of monogenic EDS types. The most recent focus was on elucidation of the pathogenetic mechanism that causes periodontal EDS (pEDS), in close collaboration with Prof. Ines Kapferer-Seebacher from the MUI Department for Operative and Restorative Dentistry. The defining clinical feature of pEDS is rapidly progressive periodontitis and tooth loss at a young age; the condition is caused by heterozygous missense or in-frame insertion/deletion mutations in the genes for complement 1 subunits C1r and C1s. Extensive functional studies of pEDS-associated C1r variants showed domain-specific abnormalities of intracellular processing and secretion with the preservation of serine protease function in the supernatant. The central elements in the pathogenesis of pEDS seem to be intracellular activation of C1r and/or C1s and extracellular presence of activated C1s independently of microbial triggers. In addition, we are involved in various studies investigating aspects of the clinical presentation of pEDS.
The institute has joined international efforts to define the different EDS types further, as a group. As the only centre from Austria and Germany, we were part of the development team for the 2017 International Classification of the Ehlers-Danlos syndromes, in which we worked primarily on the rare EDS types. The next international workshop on the rare EDS types, scheduled for May 2020 in Innsbruck but cancelled because of the COVID-19 pandemic, will now be held here in May 2021.
Neurogenetics
Sabine Rudnik
The institute is a member of the German CMT Network, which was supported by the German ministry of education and research from 2016 to 2019. Inherited peripheral neuropathies are the most common hereditary neuromuscular diseases. Within the CMT Network, we have updated the genetic diagnostic algorithms, we have studied the outcome of pregnancy and delivery in patients with CMT disease, and we are currently updating the AWMF guidelines for peripheral neuropathies in childhood.
Reproductive Medicine
Sabine Rudnik, Johannes Zschocke
In 2018 and 2019, the first guidelines were published for German-speaking countries on diagnostics and therapeutic procedures for recurrent miscarriage and on diagnosis and treatment before assisted reproduction. As part of this, we conducted a multi-centre study into andrological findings in patients with biallelic CFTR mutations causing obstructive azoospermia; for the first time, this systematically established the exact clinical and laboratory features of this condition in a large cohort.
Genetic and Genomic Counselling
Gunda Schwaninger, Johannes Zschocke, Sabine Rudnik
The rapid development of genetic diagnostic methods is producing an increased need for competent professionals who can convey the background, goals, methods, results and consequences of genetic testing in the context of individual decision-making. MSc-trained Genetic Counsellors have become widely established internationally but are still lacking in the German-speaking countries. Genetic Counsellors form a part of the interprofessional medical genetics team and complement patient care in respect of counselling aspects. In 2019, we established the first German-taught MSc programme in Genetic and Genomic Counselling at the MUI; the next 5-semester course is scheduled to start in autumn 2021. In parallel with this, we are carrying out research into the perception of this new profession by stakeholders within Austrian and German genetic services. Further information is available at http://www.gencouns.at.
European Reference Networks for Rare Diseases
Johannes Zschocke, Sabine Rudnik, Katharina Wimmer, Christine Fauth
In 2019, the institute was appointed as the Associated Centre of four European Reference Networks (ERNs): ReCONNET for connective tissue and musculoskeletal diseases, EURO-NMD for neuromuscular diseases, GENTURIS for genetic tumour risk syndromes, and ITHACA for congenital malformations and rare intellectual disability. In addition, the institute is a participator in several other Associated Centres established at the MUI and it is a co-director of the Innsbruck Centre for Rare Diseases. Many rare diseases have a primary genetic origin, and medical genetics play a major role in the diagnosis, management and research of these conditions. This requires extensive international networking. For example, within the EURO-NMD network, we published a series of patient cases with a rare subtype of nemaline myopathy caused by Austrian and Bavarian founder LMOD3 mutations. We were also able to define the genetic diagnosis in patients with unexplained myopathy, by means of exome sequencing within the MYO-SEQ consortium.
Selected Publications
- Ferreira CR, Rahman S, Keller M, Zschocke J; ICIMD advisory group. An International Classification of Inherited Metabolic Disorders (ICIMD). J Inherit Metab Dis. 2021, in press
- Koch J, Lackner K, Wohlfarter Y, Sailer S, Zschocke J, Werner ER, Watschinger K, Keller MA. Unequivocal Mapping of Molecular Ether Lipid Species by LC-MS/MS in Plasmalogen-Deficient Mice. Anal Chem. 2020;92:11268-11276
- Oemer G, Koch J, Wohlfarter Y, Alam MT, Lackner K, Sailer S, Neumann L, Lindner HH, Watschinger K, Haltmeier M, Werner ER, Zschocke J, Keller MA. Phospholipid Acyl Chain Diversity Controls the Tissue-Specific Assembly of Mitochondrial Cardiolipins. Cell Reports 2020; 30, 4281-4291.e4
- Weustenfeld M, Eidelpes R, Schmuth M, Rizzo WB, Zschocke J, Keller MA. Genotype and phenotype variability in Sjögren-Larsson syndrome. Human Mutation 2019; 40, 177–186
- Oemer G, Lackner K, Muigg K, Krumschnabel G, Watschinger K, Sailer S, Lindner H, Gnaiger E, Wortmann SB, Werner ER, Zschocke J, Keller MA. Molecular structural diversity of mitochondrial cardiolipins. Proc Natl Acad Sci U S A. 2018; 115, 4158–4163
- Wimmer K, Schamschula E, Wernstedt A, Traunfellner P, Amberger A, Zschocke J, Kroisel P, Chen Y, Callens T, Messiaen L. AG-exclusion zone revisited: Lessons to learn from 91 intronic NF1 3' splice site mutations outside the canonical AG-dinucleotides. Hum Mutat. 2020;41:1145-1156.
- Perez-Valencia JA, Gallon R, Chen Y, Koch J, Keller M, Oberhuber K, Gomes A, Zschocke J, Burn J, Jackson MS, Santibanez-Koref M, Messiaen L, Wimmer K. Constitutional mismatch repair deficiency is the diagnosis in 0.41% of pathogenic NF1/SPRED1 variant negative children suspected of sporadic neurofibromatosis type 1. Genet Med. 2020;22:2081-2088.
- Suerink M, Ripperger T, Messiaen L, Menko FH, Bourdeaut F, Colas C, Jongmans M, Goldberg Y, Nielsen M, Muleris M, van Kouwen M, Slavc I, Kratz C, Vasen HF, Brugiѐres L, Legius E, Wimmer K. Constitutional mismatch repair deficiency as a differential diagnosis of neurofibromatosis type 1: consensus guidelines for testing a child without malignancy. J Med Genet. 2019;56:53-62.
- Locher M, Steurer M, Jukic E, Keller MA, Fresser F, Ruepp C, Wöll E, Verdorfer I, Gastl G, Willenbacher W, Weger R, Nachbaur D, Wolf D, Gunsilius E, Zschocke J, Steiner N. The prognostic value of additional copies of 1q21 in multiple myeloma depends on the primary genetic event. Am J Hematol. 2021, in press.
- Malfait F, Francomano C, Byers P, … Zschocke J, Tinkle B. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175:8-26
- Rudnik-Schöneborn S, Thiele S, Walter MC, Reinecke L, Sereda M, Schöneborn R, Elbracht M. Pregnancy outcome in Charcot-Marie-Tooth disease: results of the CMT-NET cohort study in Germany. Eur J Neurol 2020;8:1390-1396
- Rudnik-Schöneborn S, Messner M, Vockel M, Wirleitner B, Pinggera GM, Witsch-Baumgartner M, Murtinger M, Kliesch S, Swoboda M, Sänger N, Zschocke J, Tüttelmann F. Andrological findings in infertile men with two (biallelic) CFTR mutations: results of a multicentre study in Germany and Austria comprising 71 patients. Hum Reprod. 2021, in press.
- Bally I, Dalonneau F, Chouquet A, Gröbner R, Amberger A, Kapferer-Seebacher I, Stoiber H, Zschocke J, Thielens NM, Rossi V, Gaboriaud C. Two Different Missense C1S Mutations, Associated to Periodontal Ehlers-Danlos Syndrome, Lead to Identical Molecular Outcomes. Front Immunol. 2019;10:2962
- Gröbner R, Kapferer-Seebacher I, Amberger A, Redolfi R, Dalonneau F, Björck E, Milnes D, Bally I, Rossi V, Thielens N, Stoiber H, Gaboriaud C, Zschocke J. C1R Mutations Trigger Constitutive Complement 1 Activation in Periodontal Ehlers-Danlos Syndrome. Front Immunol. 2019;10:2537
Selection of Funding
- Prevalence of CMMRD in NF1/SPRED1 Mutation Negatives (K Wimmer). FWF KLI 734-B26, 195k €, 2019-2021.
- Lipid peroxidation as driver of cardiolipin remodelling (M. Keller). FWF P33333, 381k €, 2020-2022
- Establishing tissue-specific cell models with culture medium additives (M. Keller). FFG Bridge project #878654, 300k €, 2020-2022
- Cellular Basis of Diseases (M. Keller). FWF doc.fund DOC-82
Collaborations
- Steven Claypool, Johns Hopkins University School of Medicine, Baltimore, USA
- José Antonio Enríquez Domínguez, Centro Nacional de Investigaciones Cardiovasculares Carlos III, Madrid, Spain
- Oroboros Instruments, Innsbruck, Austria
- Shamima Rahman, University College London, UK
- Markus Ralser, Crick Institute, London, UK
- Verena Peters, University of Heidelberg, 69117 Heidelberg, Germany
- Messiaen, Ludwine, University of Alabama at Birmingham, Birmingham, AL, USA
- Gallon, Richard, Newcastle University, Newcastle upon Tyne, UK
- Jürgen Hench, Dept. of Neropathology, Institute of Medical Genetics and Pathology, University of Basel, Switzerland
- Markus Nöthen, Institute of Human Genetics, University of Bonn, Germany
- Nicole Thielens, Christine Gaboriaud, University of Grenoble Alpes, CEA, CNRS, IBS, Grenoble, France
Devices & Services
The institute runs all relevant instruments for molecular genetic and cytogenetic analyses in an ISO 15189 accredited setting, including four massive-parallel sequencing instruments (Illumina) and the iScan array genotyping instrument (Illumina). In addition it has a timsTOF Pro mass spectrometer (Bruker) for biochemical analyses. All instruments are also available for research collaborations.
 Univ.-Prof. Dr. med. Johannes Zschocke, Ph.D.
Univ.-Prof. Dr. med. Johannes Zschocke, Ph.D.
Director
Contact:
Peter-Mayr-Str. 1
6020 Innsbruck
Austria
Email: humgen@i-med.ac.at
Phone: +43 512 9003 70500
Fax: +43 512 9003 73501
www.humgen.at